今日老師上課內容雖以microarray為主,但有許多內容仍可以和以細菌16s rRNA amplicons的高通量定序的資料處理可以互通有無。
比如廢水除氮中主要是經由硝化作用、脫硝作用以及厭氧氨氧化作用來除氮,因此廢水處理中常見的硝化菌包括Ammonium oxidizing bacteria、Nitrite oxidizing bacteria、anammox bacteria以及一些異營的denitrifier等就是會直接影響到除氮功能的族群;而某些異營菌與anammox bacteria有著nutrients如B12以及一些胺基酸交換的關係(Lawson et al., 2017),這些微生物也間接影響著整體除氮功能的表現。

回到上課老師提到的interaction→
networks→ pathways,將此概念套用至我個人的研究主題上,就可以很清晰地描繪出整個研究架構的階層,即是:從活性污泥中個別微生物間可能存在的互動關係(比如前述中AMX bacteria以及Chlorobi-affiliated bacteria間nutrients交換關係 → 以關鍵微生物為主的區域網絡 (比如隨著硝酸還原的氧化磷酸化,反應中有機物以及硝酸的來源和命運fate) → 某特定功能表現的pathways (氮的移除)。
在這樣的框架中,資料分析的處理以及邏輯扮演著非常重要的角色。雖然今日上課內容是以microarray的資料為主,但如同NGS一樣,最終都可以量化表現,因此此份作業內容我將不會一一重複上課操作的過程,主要會著重在上課教的東西我想怎樣的運用在我的研究中。
本週課程以MeV來做microarray數據的分析,data process & analysis的過程為 :
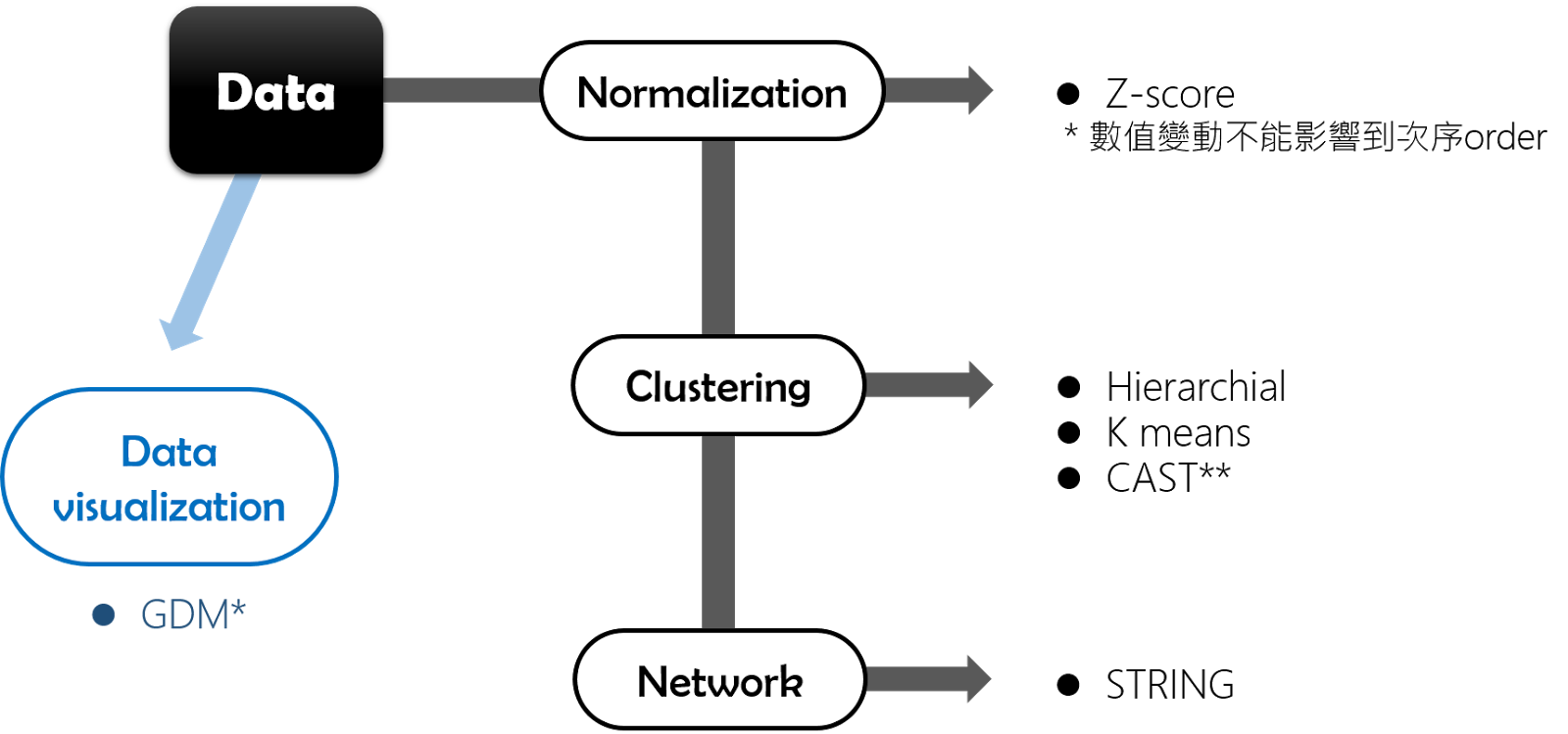
1.
檔案匯入 : 檔案格式很重要 !
File
-> Load data -> expression file loader -> select file loader ->
other format file -> 選擇要匯入檔案所在資料夾

2. Normalization
: Adjust data -> normalization -> total tensity

3. Clustering
: Hieratical clustering

分群這種資料分析的方式在環境微生物的研究中很常使用,依據各樣本菌種組成結構不同來分群,這些樣本可以是單純時間序列;可以是不同環境因子調控的實驗組,這部分在研究樣本間beta diversity時是很常用的方式。另外,也可以是依據親緣關係做分群,比如:

Distribution of sequence read abundance of
core bacterial populations (sample occupancy > 85%) at the order level (Chen, Ng, Wu, Chen,
& Wang, 2017)
比如上圖即是將各樣本中細菌reads relative abundance以bubble表示,在Y軸的分布上以親緣關係作為依據。
4.
divide genes/row by RMS
將1->5 以及 0.1-0.5這樣不同規模的變動轉換成一致的表達

做完divide genes/row by RMS後 :

5.
Visualization
-> gene distance matrix
這種用heatmap來表示樣本間相似度的關係的方法在研究環境因子的變動造成的影響時很常出現此種表示方法,因為可以很清楚的看出各種不同實驗組整體菌群結構上的差異性。另外也很常以各菌種在不同樣本中的relative abundance數據來做distance matrix,來看在各sample中微生物的變動。

這堂課學到了除了軟體的使用外更是學到了許多data process and analysis的邏輯,在不斷思考老師授課過程中講授的例子來與自己的研究領域做對照的過程,對將來要處理自己的序列資料時做基礎,不會對著滿滿的sequence不知道做完taxonomy後要幹嘛。
Reference
Chen, W. Y., Ng, T. H., Wu, J. H., Chen, J. W., & Wang, H. C.
(2017). Microbiome Dynamics in a Shrimp Grow-out Pond with Possible Outbreak of
Acute Hepatopancreatic Necrosis Disease. Sci
Rep, 7(1), 9395. doi:10.1038/s41598-017-09923-6
Lawson, C. E., Wu, S., Bhattacharjee, A. S., Hamilton, J. J.,
McMahon, K. D., Goel, R., & Noguera, D. R. (2017). Metabolic network
analysis reveals microbial community interactions in anammox granules. Nat Commun, 8, 15416.
doi:10.1038/ncomms15416
留言
張貼留言